基础教程¶
主要讲述VMD常见的命令操作。
注解
1.本节教程是有顺序的。 2.建议先阅读入门教程,再阅读基础教程。 3.所有的命令都支持tab键补全,提高效率。 4. VMD 有2个命令行窗口,一个是VMD的启动的启动窗口,一个是Extensions->TK console窗口。 5. 推荐使用 TK console窗口。 6. VMD 命令本质上是TCL language。
命令输入窗口¶
VMD 有2个命令行窗口,一个是VMD的启动的启动窗口,一个是Extensions->TK console窗口。 推荐使用 TK console窗口。
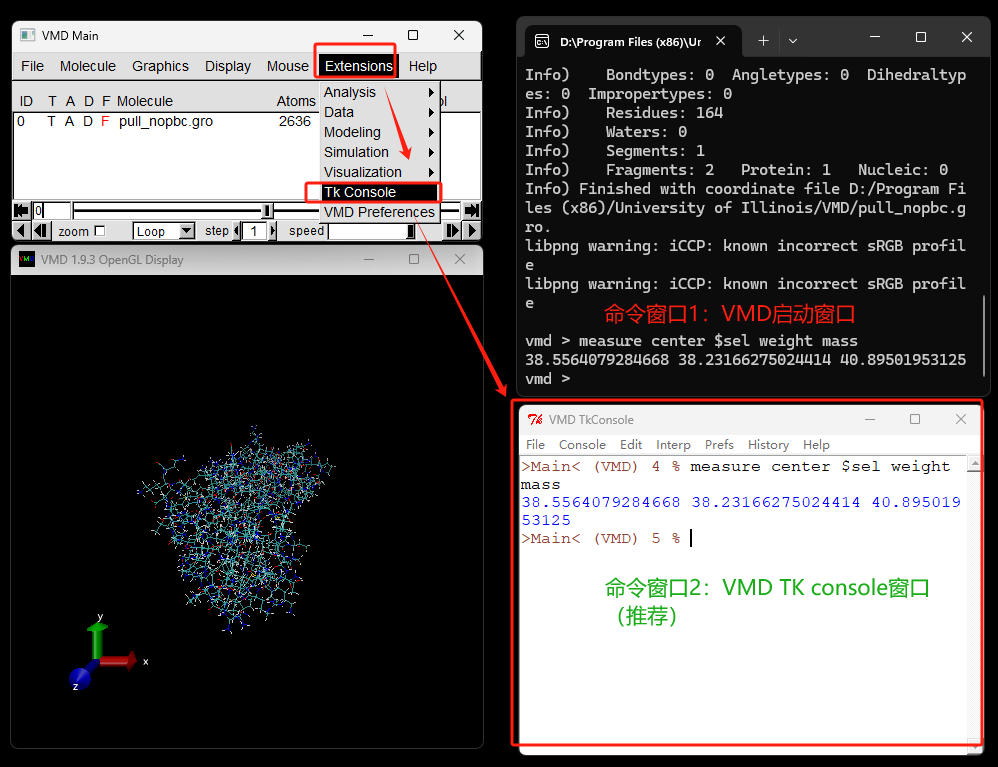
注解
注意可以把VMD当成一个TCL的模块,这样也可以在TCL脚本中调用所有VMD功能。
注解
所见即所得是VMD的一个特色,所有鼠标操作都可以录制成命令。 参见VMD高级教程 `宏录制 < >`_ 。
注解
输入命令,请用英文输入法;
忘记命令的名称¶
注解
google 关键词“vmd command measure”
忘记命令的用法¶
注解
方法一: 输入 “cmdname” 比如 “measure”, “measure center” 方法二:google 关键词“vmd measure”
笔者倾向于使用命令框TK console,下面我将讲解一些常用的命令:
查看切换工作路径(pwd)¶
默认打开和保存文件,都是在工作路径中。因此设定合适的工作路径,可以提供效率。
打开并启动VMD软件,查看和修改工作路径。
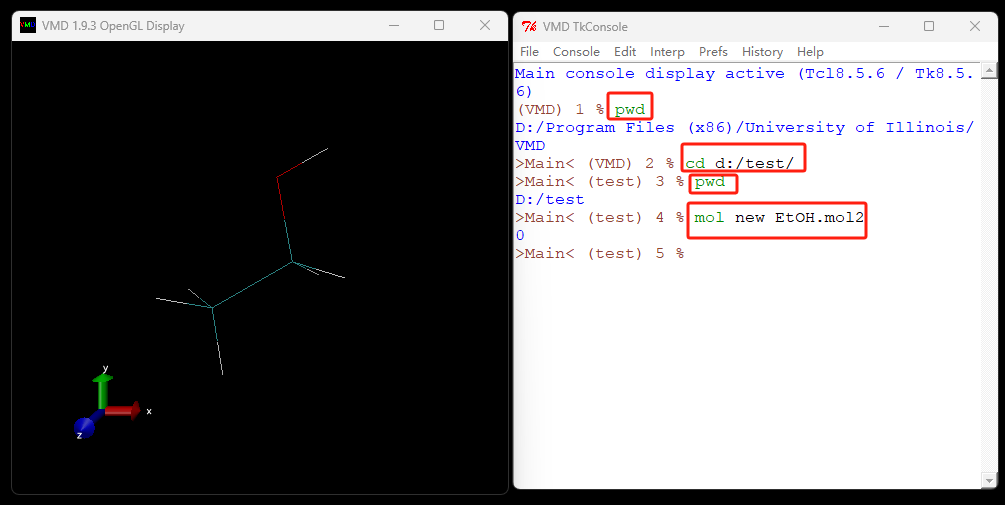
假设 D:/test 文件夹下有一个EtOH.mol2 文件,下面演示笔者喜欢的操作。
- 命令 pwd; 查看工作路径;
- 命令 cd D:/test; 切换工作路径到D盘下面的test文件夹。在执行命令前,确保D盘下面有test 文件夹。
- 命令 pwd; 查看工作路径;
- 命令 mol new; 加载分子;
如图所示:
加载分子 mol new¶
这里以 ** PDB ID: 1OWY ** 为例介绍,
1OWY.pdb 存放在 D:/test 目录下面。
>Main< (test) 15 % pwd
D:/test
>Main< (test) 16 % mol new 1OWY.pdb
8
>Main< (test) 17 % mol new 1owy.pdb
9
注解
在windowns系统上文件名称不区分大小写

计算蛋白的中心 measure center¶
这里以 ** PDB ID: 1OWY ** 为例介绍,
计算蛋白的几何中心 measure center $sel
>Main< (test) 27 % mol new 1owy.pdb
11
>Main< (test) 28 % set sel [atomselect top "protein"]
atomselect1
>Main< (test) 29 % measure center $sel
34.85237121582031 11.393967628479004 9.56042766571045
计算蛋白的质心 measure center $sel weight mass
>Main< (test) 27 % mol new 1owy.pdb
11
>Main< (test) 28 % set sel [atomselect top "protein"]
atomselect1
>Main< (test) 30 % measure center $sel weight mass
34.85234451293945 11.385374069213867 9.555062294006348
>Main< (test) 31 %
计算配体的中心 measure center¶
核心命令是 measure center 。
关键是 atomelect语法,选择感兴趣的配体原子 。
计算配体的几何中心
>Main< (test) 38 % mol new 1owy.pdb
13
>Main< (test) 39 % set ligPRY [ atomselect top "resname PRY" ]
atomselect3
>Main< (test) 40 % measure center $ligPRY
27.018299102783203 7.100600719451904 3.5752999782562256
计算配体的质心
>Main< (test) 38 % mol new 1owy.pdb
13
>Main< (test) 39 % set ligPRY [ atomselect top "resname PRY" ]
atomselect3
>Main< (test) 41 % measure center $ligPRY weight mass
27.041915893554688 7.121399402618408 3.5915112495422363
>Main< (test) 42 %
删除分子 mol delete¶
删除所有加载的分子
mol delete all
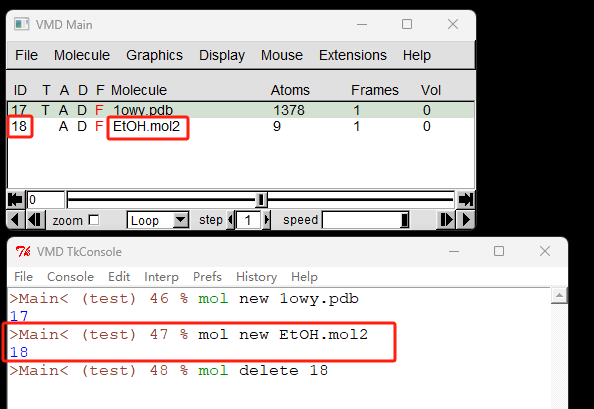
基于分子ID,删除不想要的分子,如下图所示,删除EtOH分子。
step1. 在分子列表窗口查看EtOH对应的分子ID是18;另外加载分子的时候,也会返回分子ID;
step2. mol delete 18
查找氨基酸残基的信息¶
查找110号氨基酸CA原子编号
set sel [atomselect top "resid 110 and name CA"]
$sel get index
查找110号氨基酸残基的名称
set sel [atomselect top "resid 110 and name CA"]
$sel get resname
