高级教程¶
主要讲述VMD中脚本的编写、命令的自定义、插件的编写、插件的介绍。
脚本、命令、插件¶
VMD支持TCL 编程语言,故可以借助TCL让VMD无所不能,VMD除了显示软件,也能成为计算软件。
脚本:把VMD作为一个模块,利用已有的命令实现一些计算功能。
命令:VMD中内置了一些命令,如measure等,我们也可以自定义新的命令。
插件:VMD中内置了一些插件,如MovieMaker等,我们也可以自定义插件。
注解
编写VMD脚本、命令、插件,需要有一定的编程基础就可以。
Tcl (Tool Command Language)脚本语法¶
Tcl 的核心哲学与 Python 完全不同:在 Tcl 中,“一切皆字符串”,并且所有操作都是命令。
赋值
python:
name = 520
Tcl:
set name 520
在TCL中赋值是通过命令set命令完成的,且在特定命令环境下自动区分数字还是字符串。
输出值
print(name)
Tcl:
puts $name
通过在变量名前面加$符号,实现把变量名换成其对应的值。
替换机制 (Substitution)
- 变量替换 ($):把变量名换成它的值。
- 命令替换 ([]):类似于 Python 的函数调用返回值。
数据结构 伪列表
Tcl 没有 Python 那么丰富的内置类型,它只有伪装成字符串的List。
set mylist1 {protein water ions}
set mylist2 "protein water ions"
控制流 (Control Flow) if
if {$a > 10} {
puts "Big"
} else {
puts "Small"
}
控制流 (Control Flow) foreach
set atoms {CA CB N O}
foreach atom $atoms {
puts "Atom type: $atom"
}
定义函数
# proc 函数名 {参数列表} {函数体}
proc calculate_dist {x1 y1 z1} {
set dist [expr sqrt($x1**2 + $y1**2 + $z1**2)]
return $dist
}
# 调用
set d [calculate_dist 1 2 3]
读文件
set in [open "test.txt" r]
set content [read $in]
close $in
puts $content
写文件
# Python: f = open("test.txt", "w")
set out [open "test.txt" w]
# Python: f.write("Hello VMD\n")
puts $out "Hello VMD"
puts $out "Line 2: Everything is a string."
# Python: f.close()
close $out
注解
“”(双引号):允许内部进行变量替换 ($) 和命令替换 ([])。
{}(大括号):禁止一切替换。内部是什么样,存进去就是什么样
脚本1 对齐和移动2个构象¶
假设2个构象对应的分子ID分别是0和1。 通过如下脚本命令把1的构象移动到0的构象上。
set ref [atomselect 0 "backbone"]
set target [atomselect 1 "backbone"]
measure fit $target $ref
$target move [measure fit $target $ref]
安装VMD中的命令¶
这里我以这个命令 `FocalBlur<https://pymolwiki.org/index.php/FocalBlur>`_ 为例进行演示。
- 下载 focal_blur.py脚本,https://raw.githubusercontent.com/Pymol-Scripts/Pymol-script-repo/master/focal_blur.py 比如下载到D盘根目录下面 d:/focal_blur.py
- 打开pymol,在命令窗口输入run d:/focal_blur.py 就完成了安装。
- 运行命令 FocalBlur aperture=4,samples=400,ray=0
FocalBlur一共有5个参数:
- aperture
- samples
- ray
- width
- height
该命名和单反的光圈相关,控制景深的。 需要开启ray=1 才能看到光圈景深的效果
修改VMD的默认设置¶
我个人习惯保存突变的时候使用白色背景、关闭光照阴影,而默认的是透明背景, 每次保存图片的时候都需要用命令” set ray_opaque_background, 1 “设置白色背景,比较麻烦。 我们可以通过下面的方法,修改PyMOL的默认设置。
建议创建一个脚本 pymolset.py, 假设该脚本存放在D盘
# AUTOGENERATED FILE
from pymol import cmd, invocation
cmd.set("bg_rgb", 'white')
cmd.set("ray_opaque_background", '1')
cmd.set("ray_shadow", 'off')
如果不知道如何定义pymolset.py,可以通过 save_settings.py
如果无法下载从国外网站下载,可通过下面的百度云链接进行下载。
链接:https://pan.baidu.com/s/1vWNU5ssyOTvMkPAHVqQ27g?pwd=ejsg
提取码:ejsg
在PyMol 命令窗口下,先修改成自己想要的设置,运行下面的命令就可以自动生成pymolset.py 文件。
run save_settings.py
save_settings pymolset.py
生成pymolset.py后,用文本编辑器打开进行编辑修改,保留自己需要设置就可以了。
创建命令¶
这里我举一个例子: 计算两个苯环之间的距离或者五元环和苯环之间的距离。
拓展:计算2个object的中心距离。
首先我们为命令起名字,要求是简洁高效,通过命令就能猜测到这个命令的功能。 这里我起的脚本名 dist_center。 核心是在python 脚本中定义这个函数,并cmd.extend拓展命令。 框架如下
1 2 3 4 | from pymol import cmd
def dist_center(obj1="obj01",obj2="obj02",showflag=True):
pass
cmd.extend("dist_center", dist_center)
|
完整代码(dist_center_2obj.py)如下:
演示如下:
载入蛋白,运行run dist_center_2obj.py命令; 把TRY-72侧链苯环6个原子保存为obj01; 把HIS-351侧链咪唑环的5个原子保存为obj02; 运行命令 dist_center obj01,obj02 即可测量距离。

创建向量draw_vector¶
VMD的宏录制¶
点击 File->Log TCL COMMAND TO FILE -> 填写宏的名字 xxx.txt 然后操作VMD, 操作完成后点击 File->Trun off logging;
文本编辑器(如vscode)打开宏文件就可以查看里面的命令。
VMD插件fftk推荐¶
fftk介绍¶
fftk (Force Field Toolkit) 是一个用于力场参数优化和生成的 VMD 插件, 旨在简化和自动化分子模拟中力场参数的生成和调整过程。
fftk安装¶
fftk是VMD内置的插件,安装VMD后就自动包含该插件,无需额外安装。
fftk 依赖软件¶
- Gaussian软件,对软件版本要求较高,部分版本的Gaussian可能无法计算Z-matrix 格式的文件。
注解
建议使用正版g09 Gaussian,出现问题和官方联系; 或者使用g16版本的Gaussian, 修改log文件格式保持和g019一致。 fftk 是基于g09开发的。
- namd2/namd3, NAMD和VMD都是由伊利诺伊大学香槟分校的理论与计算生物物理研究组开发。
fftk优化流程¶
文献:
C.G. Mayne, J. Saam, K. Schulten, E. Tajkhorshid, J.C. Gumbart. J. Comput. Chem. 2013, 34, 2757-2770.
fftk 优化流程如下图所示:
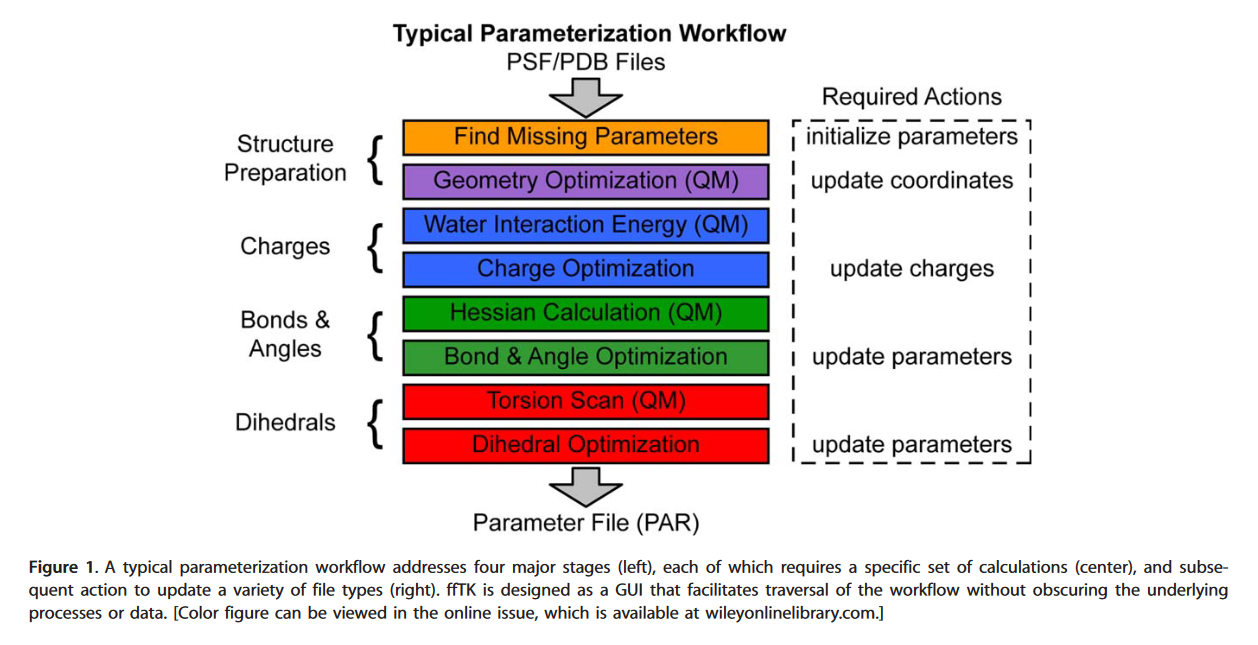
首先准备小分子的PDB文件和小分子的PSF文件, 可通过插件MoleFacture插件生成。
PSF文件:是分子的拓扑结构文件,包含体系所有分子的键、角、二面角、非正常二面角等拓扑信息。
NAMD 模拟,通常需要 4 个文件:
1. PDB 文件:包含系统的原子坐标信息。
2. PSF 文件:包含系统的拓扑信息。
3. PRM 文件:包含力场参数信息。
4. 配置文件(Conf 文件):定义模拟控制参数。
然后是结构准备, 接着是电荷参数优化、键和角度参数优化、二面角优化, 最后是转换成Gromcs等动力学软件的格式。
step0 从头开始准备pdb 和psf 文件¶
注解
从头准备pdb 和psf文件适合简单的片段小分子(MW<300), 对于复杂小分子(MW>300)推荐在cgenff的基础上进行优化参数。
官方教程是以乙醇EtOH分子为例进行介绍fftk操作的。 分子越简单,遇到的问题越少,可以先尝试简单分子熟悉操作。 再用fftk 应用到自己需要的分子上。
这里我以`Iptacopan <https://files.rcsb.org/ligands/download/JGQ_ideal.sdf>`_ 分子为例进行介绍,
首先转换成JGQ.mol2 文件,这是一个两性分子,包含一个质子和一个羧酸;修该mol2中的残基名称为JGQ 然后提交到 cgenff 网站处理; cgenff网站会基于结构给出罚分,如下图所示:
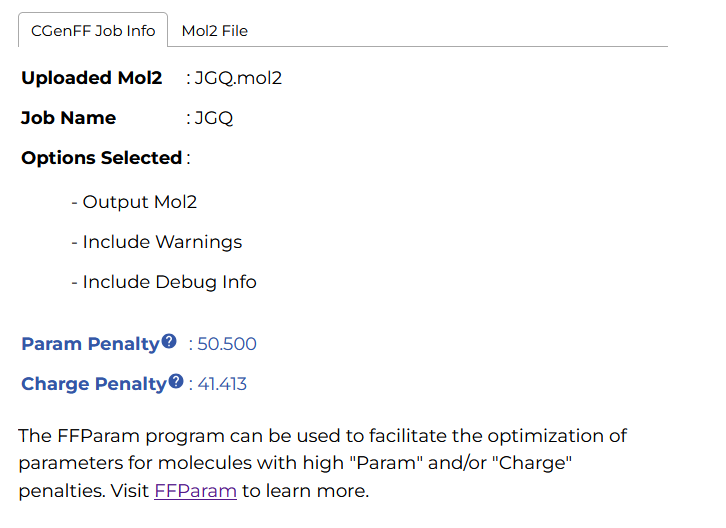
我们看Param Penalty: 50.500;Charge Penalty: 41.413。Penalties 超过50需要验证和优化参数; cgenff推荐用 FFParm 工具去优化力场参数, 这里我们使用 fftk 工具进行优化,这2个工具的原理是相似的。
注解
Penalty is the highest penalty score of the associated parameters. Penalties lower than 10 indicate the analogy is fair; penalties between 10 and 50 mean some basic validation is recommended; penalties higher than 50 indicate poor analogy and mandate extensive validation/optimization.
下载 JGQ.zip 和 JGQ_gromacs.zip 文件。
准备 psf 和 pdb 文件¶
从JGQ.zip文件中获得1. JGQ.cgenff.mol2 和 2. JGQ.str;
把JGQ.cgeff.mol2文件中sybyl的原子类型替换为charmm的原子类型(来自JGQ.str文件), 保存为 JGQ.cgeff.upate.mol2 。
VMD 加载分子JGQ.cgeff.upate.mol2,然后打开插件 Extensions -> Modeling ->MoleFacture, Selection文本框中填写 all ,然后点击start MoleFacture 按钮。
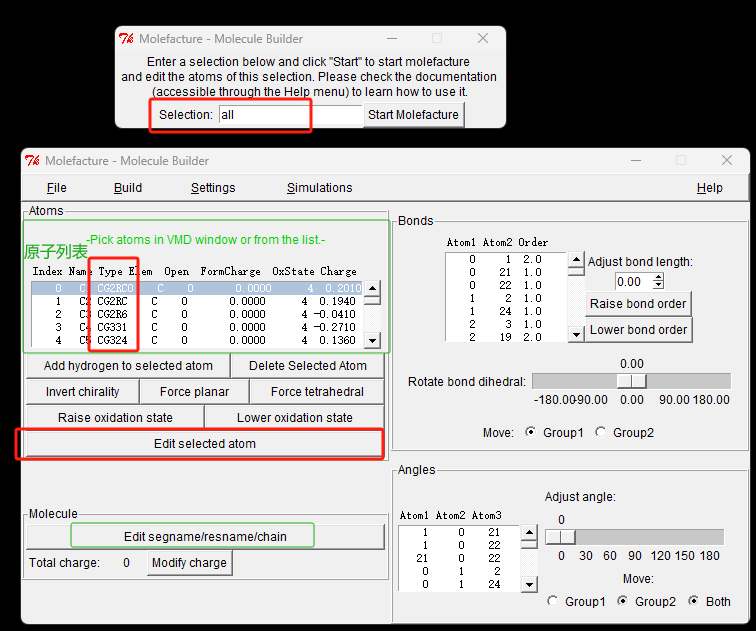
在原子列表中依次选择原子, 对照JGQ.cgeff.upate.mol2修改原子类型(mol2文件原子类型最多是5位,而charmm的原子类型最多是6位),点击Edit selcted atom 修改原子类型。
注解
原子列表中的有7个原子属性。 1. Index原子索引 2. Name 原子名称 3. Type 原子类型charmm 4. Elem 元素 5. Open 饱和状态,比如C 连4根键,则为饱和,还剩0根键可连;N连4根键,则过饱和,为-1;负电荷O,为1。 6. FormCharge 形式电荷 7. OxState 价键数目,C是4,N是3,O 是2, H是1。
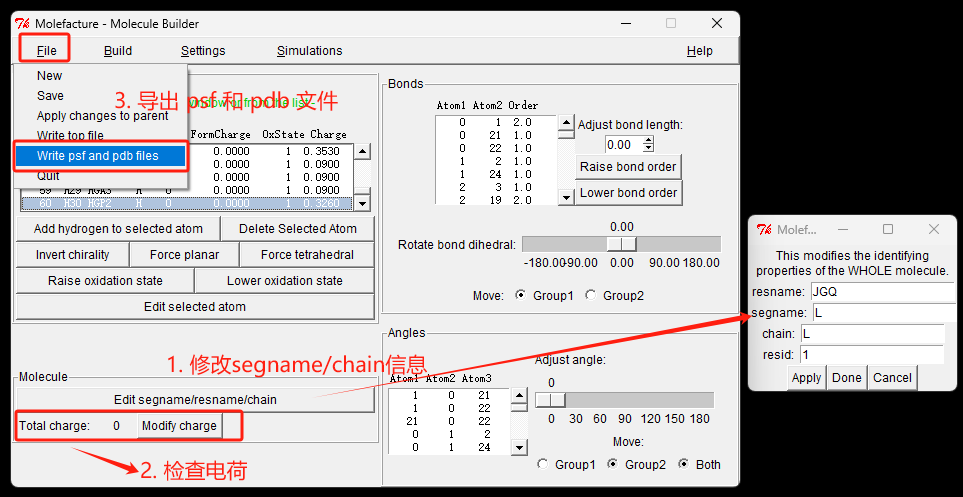
原子列表修改完成后,在分子Molecule列表中,点击Edit segname/resname/chain 按钮设置segame和chain为L。
JGQ是两性分子,净电荷为0。
最后在MoleFacture窗口中点击FiLe->write psf and pdb files 按钮,导出PSF 和 PDB 文件。 如下图所示:
step1 从头结构准备¶
首先 准备初始化的par文件;
- 点击 Extensions-> Modeling -> Force Field toolkit 工具,打开fftk插件;选择BuildPar标签页,在点开 Identify Missing parameters 内容。
- 点击Browse 加载psf 文件。
- 点击Browse 加载pdb文件。
- 点击analye 按钮;
- 点击save as 指定待保存的par 文件的位置;
- 点击Write initial parameter file, 保存par文件;
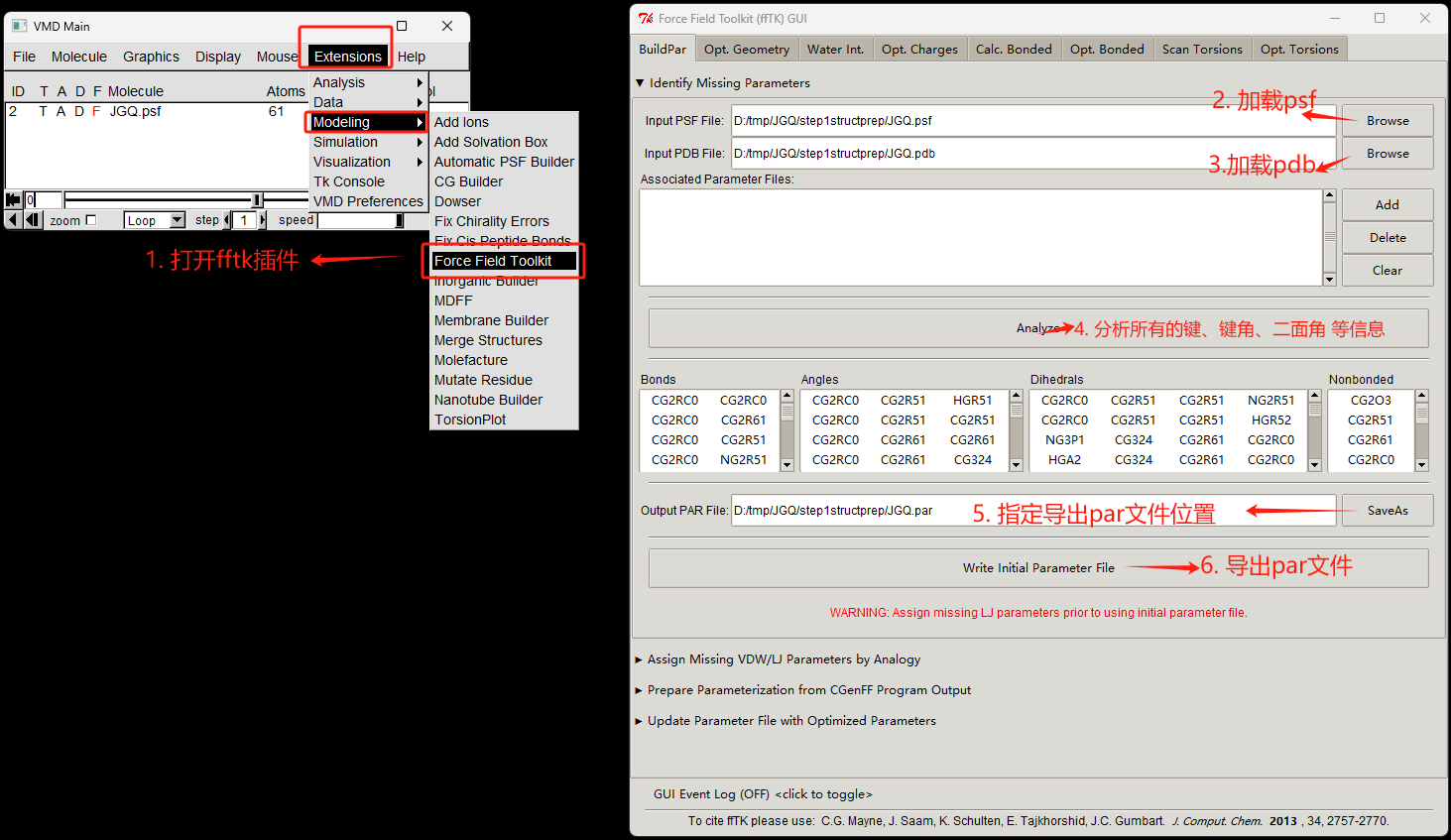
接着 给par文件增加VDW/LJ参数;
- 关闭 Identify Missing parameters ;打开 Assign Missing Parameter by analogy;
- 点击 Browse 按钮定位到par文件;
- 点击 load 加载par文件;
- 加载charmm力场文件 top_all36_cgenff.rtf 和 par_all36_cgenff.prm
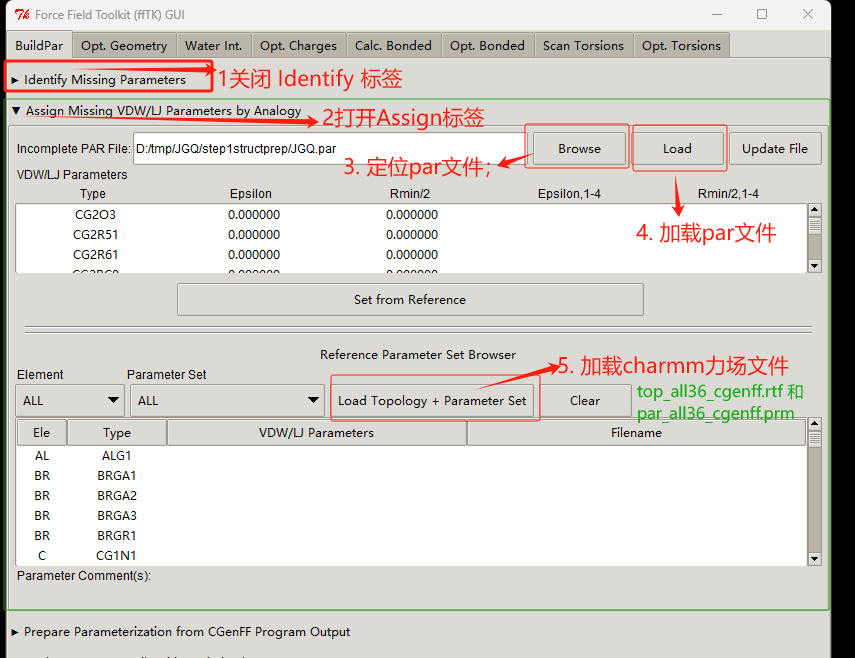
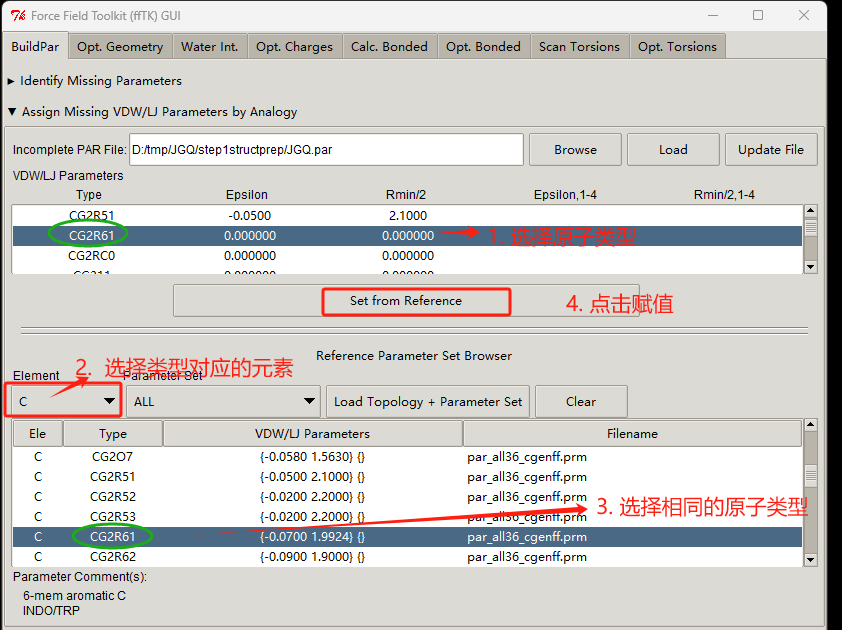
- 依次选择原子类型;
- 基于元素过滤;
- 找到对应的原子类型;
- 点击Set from reference 按钮
如下图所示:
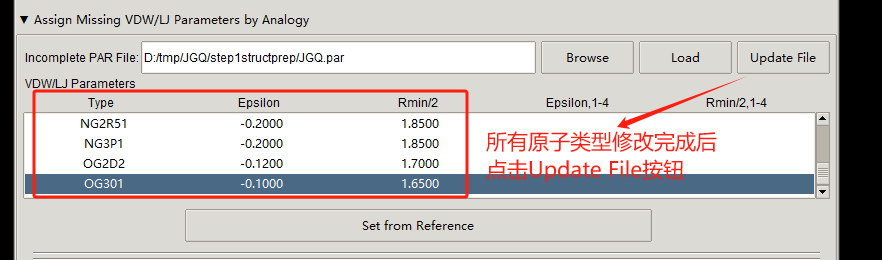
对所有原子类型进行VDW/LJ赋值以后,点击Update File 更新par 文件,如下图所示:
step1 从cgenff 网站开始准备PSF和PDB和PAR(推荐)¶
这里我以`Iptacopan <https://files.rcsb.org/ligands/download/JGQ_ideal.sdf>`_ 分子为例进行介绍,
首先转换成JGQ.mol2 文件,这是一个两性分子,包含一个质子和一个羧酸;修该mol2中的残基名称为JGQ 然后提交到 cgenff 网站处理;
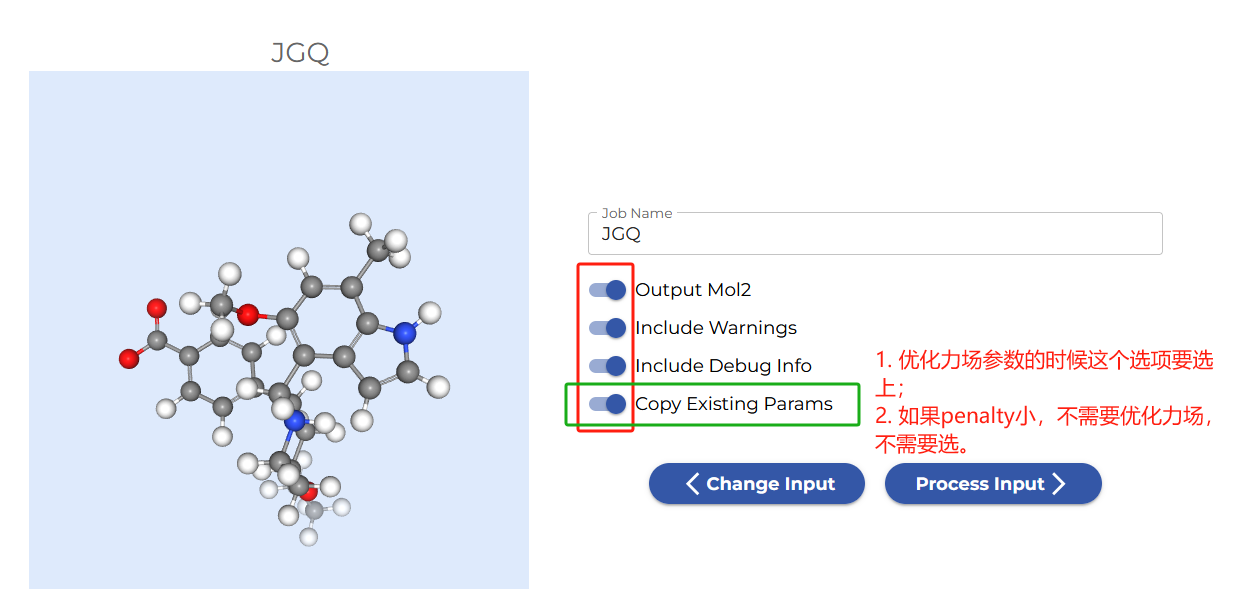
注解
一定要勾选copy existing parameter选项;
cgenff网站会基于结构给出罚分,如下图所示:
经过cgenff网站处理获得 JGQ.cgenff.mol2 和 JGQ.str。
我们看Param Penalty: 50.500;Charge Penalty: 41.413。Penalties 超过50需要验证和优化参数; cgenff推荐用 FFParm 工具去优化力场参数, 这里我们使用 fftk 工具进行优化,这2个工具的原理是相似的。
首先 从cgenff文件准备psf/pdb/par文件;
- 点击 Extensions-> Modeling -> Force Field toolkit 工具,打开fftk插件;选择BuildPar标签页,在点开 Prepare Parameterization from CGenFF Program Output 内容。
- 点击Browse 加载mol2 文件JGQ.cgenff.mol2。
- 点击Browse 加载str文件JGQ.str。
- 点击Browse 指定待保存的psf/pdb/par 文件的位置;
- 设置resname,segname,chain信息;
- 点击Analyze Input按钮找到异常参数;
- 点击write psf/pdb 按钮生成JGQ.psf 和JGQ.pdb文件;
- 点击write par 按钮生成JGQ.analogy.par 和JGQ.existing.par 文件;
JGQ.analogy.par 是基于相似性的从charmm36中推测的参数,可以作为起点继续优化,尤其是对penalty比较高的参数进行优化。 JGQ.existing.par 是包含在charmm36中的力场参数,通常具有较高的准确性和可靠性,如果文件中没有参数, 说明cgenff网站计算的时候没有勾选copy existing parameters参数。
合并JGQ.existing.par 和JGQ.analogy.par 为 JGQ.par。 同时加上原子类型的vdw 和LJ 参数。 至此完成了psf/pdb/par的准备工作。
这里放一个初始化0的par 文件: JGQ.ini.par 。
!=============================================================
!
! Parameter file generated by the Force Field ToolKit (ffTK)
!
! For additional information, see:
! http://www.ks.uiuc.edu/Research/vmd/plugins/fftk
! http://www.ks.uiuc.edu/Research/fftk
!
! Authors:
! Christopher G. Mayne
! Beckman Institute for Advanced Science and Technology
! University of Illinois, Urbana-Champaign
! http://www.ks.uiuc.edu/~mayne
! mayne@ks.uiuc.edu
!
! James C. Gumbart
! Georgia Institute of Technology
! http://simbac.gatech.edu
! gumbart_physics.gatech.edu
!
! If you use parameters developed using ffTK, please cite:
! C.G. Mayne, J. Saam, K. Schulten, E. Tajkhorshid, J.C. Gumbart. J. Comput. Chem. 2013, 34, 2757-2770.
!
!=============================================================
BONDS
!V(bond) = Kb(b - b0)**2
!
!Kb: kcal/mole/A**2
!b0: A
!
!atom type Kb b0
!
CG2RC0 CG2RC0 0.000 0.000 !
CG2RC0 CG2R61 0.000 0.000 !
ANGLES
!
!V(angle) = Ktheta(Theta - Theta0)**2
!
!V(Urey-Bradley) = Kub(S - S0)**2
!
!Ktheta: kcal/mole/rad**2
!Theta0: degrees
!Kub: kcal/mole/A**2 (Urey-Bradley)
!S0: A
!
!atom types Ktheta Theta0 Kub S0
!
!
CG2RC0 CG2R51 HGR51 0.000 0.000 !
CG2RC0 CG2R51 CG2R51 0.000 0.000 !
DIHEDRALS
!
!V(dihedral) = Kchi(1 + cos(n(chi) - delta))
!
!Kchi: kcal/mole
!n: multiplicity
!delta: degrees
!
!atom types Kchi n delta
!
CG2RC0 CG2R51 CG2R51 NG2R51 0.0000 1 0.00 !
CG2RC0 CG2R51 CG2R51 HGR52 0.0000 1 0.00 !
IMPROPER
!
!V(improper) = Kpsi(psi - psi0)**2
!
!Kpsi: kcal/mole/rad**2
!psi0: degrees
!note that the second column of numbers (0) is ignored
!
!atom types Kpsi psi0
!
NONBONDED nbxmod 5 atom cdiel shift vatom vdistance vswitch -
cutnb 14.0 ctofnb 12.0 ctonnb 10.0 eps 1.0 e14fac 1.0 wmin 1.5
!
!V(Lennard-Jones) = Eps,i,j[(Rmin,i,j/ri,j)**12 - 2(Rmin,i,j/ri,j)**6]
!
!epsilon: kcal/mole, Eps,i,j = sqrt(eps,i * eps,j)
!Rmin/2: A, Rmin,i,j = Rmin/2,i + Rmin/2,j
!
!atom ignored epsilon Rmin/2 ignored eps,1-4 Rmin/2,1-4
!
CG2O3 0.0 0.000000 0.000000 ! ! SET BY ANALOGY!!!
CG2R51 0.0 0.000000 0.000000 ! ! SET BY ANALOGY!!!
END
和一个cgenff赋值后的par 文件:`JGQ.cgenff.par < https://pan.baidu.com/s/17fyQ-2am3F1UV5PtAMbTUA?pwd=1scx>`_ 。
!=============================================================
!
! Parameter file generated by the Force Field ToolKit (ffTK)
!
! For additional information, see:
! http://www.ks.uiuc.edu/Research/vmd/plugins/fftk
! http://www.ks.uiuc.edu/Research/fftk
!
! Authors:
! Christopher G. Mayne
! Beckman Institute for Advanced Science and Technology
! University of Illinois, Urbana-Champaign
! http://www.ks.uiuc.edu/~mayne
! mayne@ks.uiuc.edu
!
! James C. Gumbart
! Georgia Institute of Technology
! http://simbac.gatech.edu
! gumbart_physics.gatech.edu
!
! If you use parameters developed using ffTK, please cite:
! C.G. Mayne, J. Saam, K. Schulten, E. Tajkhorshid, J.C. Gumbart. J. Comput. Chem. 2013, 34, 2757-2770.
!
!=============================================================
BONDS
!V(bond) = Kb(b - b0)**2
!
!Kb: kcal/mole/A**2
!b0: A
!
!atom type Kb b0
!
CG2O3 CG2R61 200.000 1.500 ! 3CPY, pyridine-3-carboxylate (PYRIDINE nicotinic acid), yin
CG2O3 OG2D2 525.000 1.260 ! PROT adm jr. 7/23/91, acetic acid
ANGLES
!
!V(angle) = Ktheta(Theta - Theta0)**2
!
!V(Urey-Bradley) = Kub(S - S0)**2
!
!Ktheta: kcal/mole/rad**2
!Theta0: degrees
!Kub: kcal/mole/A**2 (Urey-Bradley)
!S0: A
!
!atom types Ktheta Theta0 Kub S0
!
!
CG2R61 CG2O3 OG2D2 40.000 116.000 50.00 2.3530 ! 3CPY, pyridine-3-carboxylate (PYRIDINE nicotinic acid), yin
OG2D2 CG2O3 OG2D2 100.000 128.000 70.00 2.2587 ! PROT adm jr. 7/23/91, correction, ACETATE (KK)
DIHEDRALS
!
!V(dihedral) = Kchi(1 + cos(n(chi) - delta))
!
!Kchi: kcal/mole
!n: multiplicity
!delta: degrees
!
!atom types Kchi n delta
!
OG2D2 CG2O3 CG2R61 CG2R61 3.1000 2 180.00 ! BIPHENYL ANALOGS, peml
CG2RC0 CG2R51 CG2R51 NG2R51 4.0000 2 180.00 ! PROT JWK 05/14/91 fit to indole
IMPROPER
!
!V(improper) = Kpsi(psi - psi0)**2
!
!Kpsi: kcal/mole/rad**2
!psi0: degrees
!note that the second column of numbers (0) is ignored
!
!atom types Kpsi psi0
!
CG2O3 OG2D2 OG2D2 CG2R61 96.0000 0.00 ! PROT 90.0->96.0 acetate, single impr (KK) WILDCARD
NONBONDED nbxmod 5 atom cdiel shift vatom vdistance vswitch -
cutnb 14.0 ctofnb 12.0 ctonnb 10.0 eps 1.0 e14fac 1.0 wmin 1.5
!
!V(Lennard-Jones) = Eps,i,j[(Rmin,i,j/ri,j)**12 - 2(Rmin,i,j/ri,j)**6]
!
!epsilon: kcal/mole, Eps,i,j = sqrt(eps,i * eps,j)
!Rmin/2: A, Rmin,i,j = Rmin/2,i + Rmin/2,j
!
!atom ignored epsilon Rmin/2 ignored eps,1-4 Rmin/2,1-4
!
CG2O3 0.0 -0.070000 2.000000 !
CG2R51 0.0 -0.050000 2.100000 !
END
step2 结构优化¶
首先 准备Gaussian输入文件;
- 点击 Extensions-> Modeling -> Force Field toolkit 工具,打开fftk插件;选择Opt.Geometry标签页。
- 点击Browse 加载pdb 文件JGQ.pdb。
- 点击Browse 设置Gaussian输入文件的位置JGQ.opt.gau。
- 设置Gaussian输入文件参数。
- 点击write Gaussian input file 按钮,生成JGQ.opt.gau 文件

然后 利用量化软件Gaussian对结构进行优化
在终端运行下述命令:
nohup g16 JGQ.opt.gau &
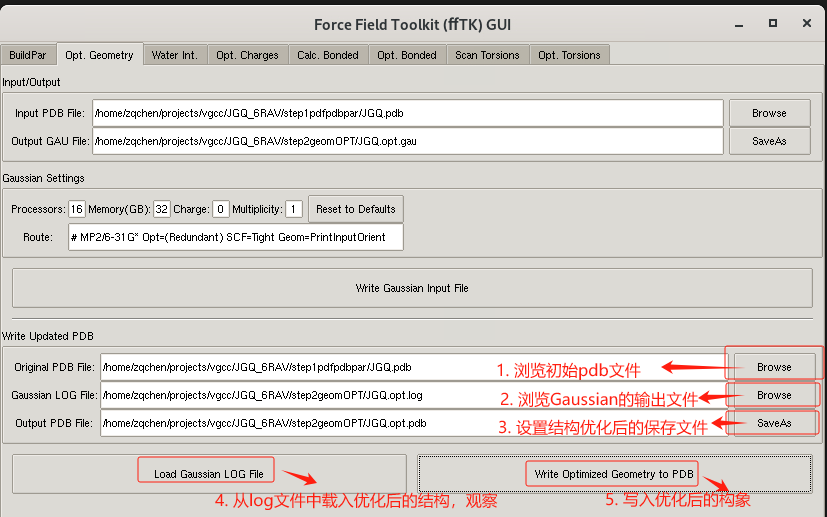
最后 输出优化后的结构
- 点击Browse 加载pdb 文件JGQ.pdb。
- 点击Browse 设置Gaussian输出文件的位置JGQ.opt.log。
- 点击save as 设置优化后的结构文件JGQ.opt.pdb。
- 点击load Gaussian log fle 加载优化后结构到VMD窗口中;检查结构是否正确,比如H原子的位置。
- 若结构没有问题,点击write Optimized Geometry to PDB 按钮,生成JGQ.opt.pdb 文件。若结构有问题,修改结构或者参数重新优化。
如下图所示:
step3 水相互作用能计算watint¶
首先 准备计算配体和水分子相互作用能的Gaussian输入文件
注解
计算配体和水分子相互作用能需要3个文件:1. 复合物文件;3.配体文件;4.水分子文件;
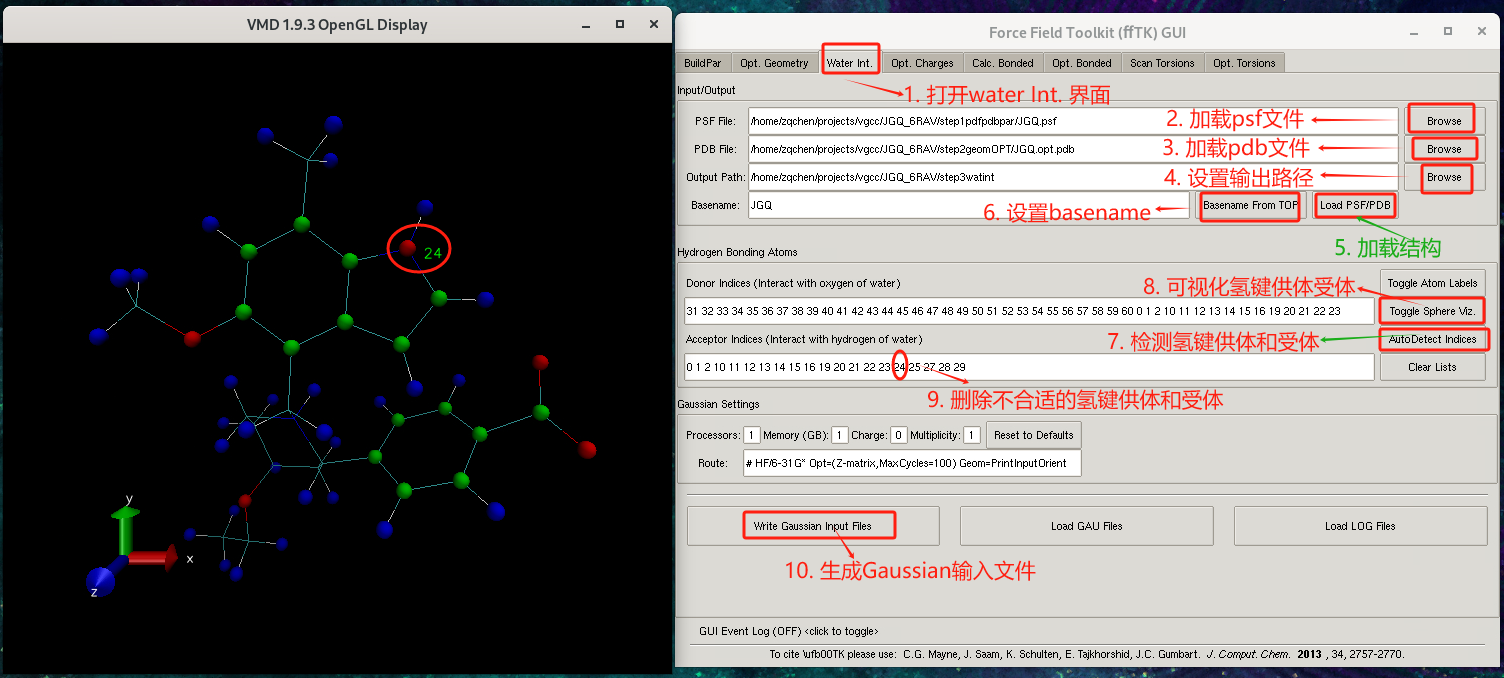
点击 Extensions-> Modeling -> Force Field toolkit 工具,打开fftk插件;选择Water Int.标签页。
点击Browse 加载 psf 文件JGQ.psf。
点击Browse 加载结构优化后的 pdb 文件JGQ.opt.pdb。
设置Water Int. 工作路径,计算water int.所有输出文件的路径。
点击load psf/pdb 加载结构信息到VMD中。
点击Base name from top 设置Basename。
点击write Gaussian input file 按钮,生成JGQ.opt.gau 文件
点击 AutoDetect Indices 按钮自动检测配体上的氢键受体和氢键供体;只有氢键供体和氢键受体才可以和水分子形成稳定相互作用;
点击 Toggle Sphere Viz. 确认氢键供体和受体是否正确。
- 红色球:表示氢键受体。
- 蓝色球:表示氢键供体。
- 绿色球:表示同时为氢键供体和受体。
吲哚的 N 原子的索引为 24 号,被标记为红色小球(氢键受体),但由于 N 原子的孤对电子与芳香环共轭,通常不作为氢键受体。 因此,需要从 Acceptor Indices中移除索引 24。 碳原子被标记为绿色,因此应移除碳原子的索引范围:0-2, 19-24, 10-16。
检查完成后,设置Gaussian参数,并点击Write Gaussian Input Files生成Gaussian的输入文件。
如下图所示:
注解
注意:不是所有的氢键受体和供体附近都适合放置水分子的。 检查所有Gau输入文件中水分子位置是否合适,如果不合适,手动调整到合适的位置, 如果无法调整到合适的位置则删除文件。
然后 执行Gaussian计算
nohup g16 xxx.gau &
或者
nohup g09 xxx.gau &
最后 检出Gaussian输出结构是否合理
在VMD 或者 gview中检查输出结构,如果不合适,手动调整到合适的位置,重新优化。 如果无法调整到合适的位置则删除文件。
至此完成了所有的准备工作。
step4 电荷优化更新psf文件¶
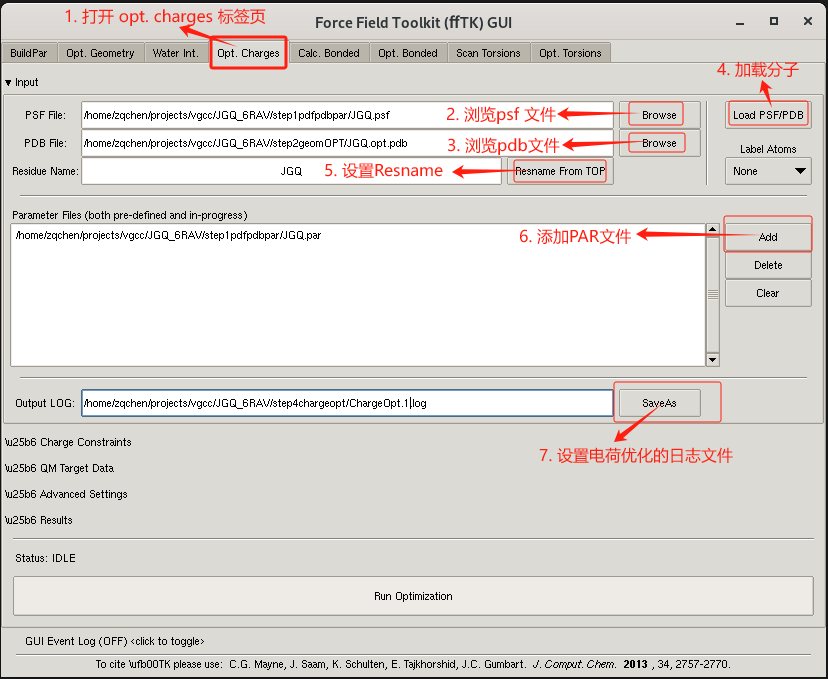
首先 准备电荷优化的输入文件;
- 点击 Extensions-> Modeling -> Force Field toolkit 工具,打开fftk插件;选择Opt.charges标签页,展开input的内容。
- 点击Browse 加载psf 文件JGQ.psf。
- 点击Browse 加载pdb 文件 JGQ.opt.pdb。
- 点击load psf/pdb按钮加载分子到VMD中。
- 点击Resname from top 设置Resname。
- 点击add 按钮加载par 文件JGQ.par。
- 点击save as 按钮,设置电荷优化过程中的日志文件,建议文件名的命名方式ChargeOpt.n.log。n从0开始,每优化一次加1。
如下图所示:
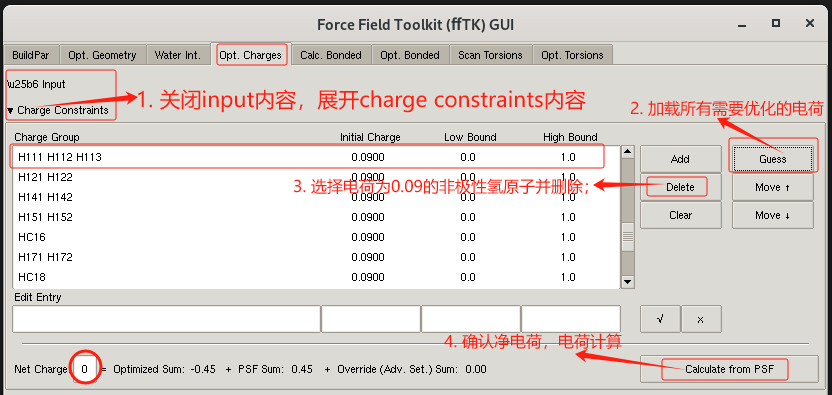
然后 设置需要优化的电荷
- 关闭input的内容,打开charge constraints内容。
- 点击Guess 加载所有的原子电荷。
- 依次选中电荷为+0.09的非极性氢原子并删除。迭代循环,删除所有的电荷为+0.09的非极性氢原子。这是charmm力场固定的电荷,不需要优化。
- 确认分子的净电荷,并点击Calculate from psf按钮进行计算,需要优化的电荷总和。
如下图所示:
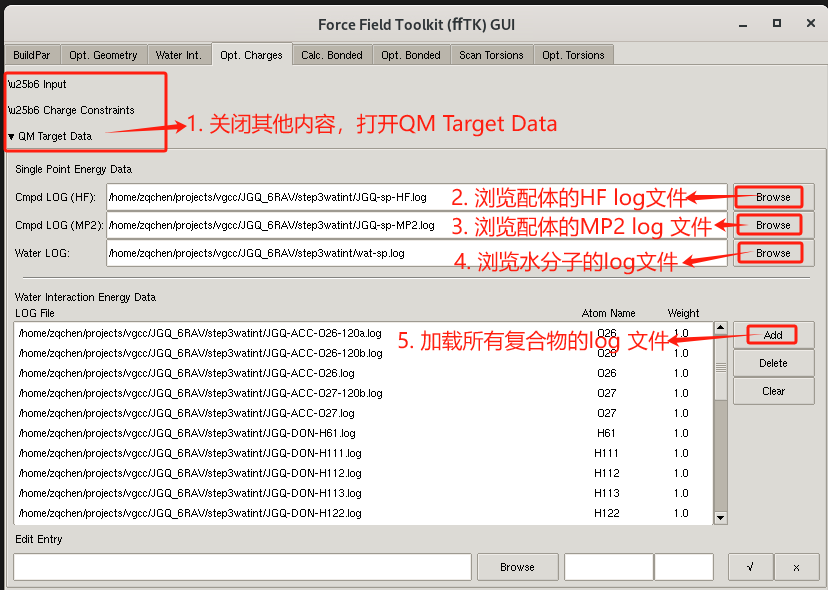
接着 载入量化的计算结果
- 关闭charge constraints内容,打开QM Target Data内容。
- 浏览配体的HF2 log文件 JGQ-sp-HF2.log。
- 浏览配体的MP2 log文件 JGQ-sp-MP2.log。
- 浏览水分子的HF2 log文件 wat-sp.log。
- 点击add 按钮,加载所有配体和水分子复合物的log文件。
如下图所示:
注解
高级设置针对力场开发者,这里暂不介绍。采用默认设置就可以。
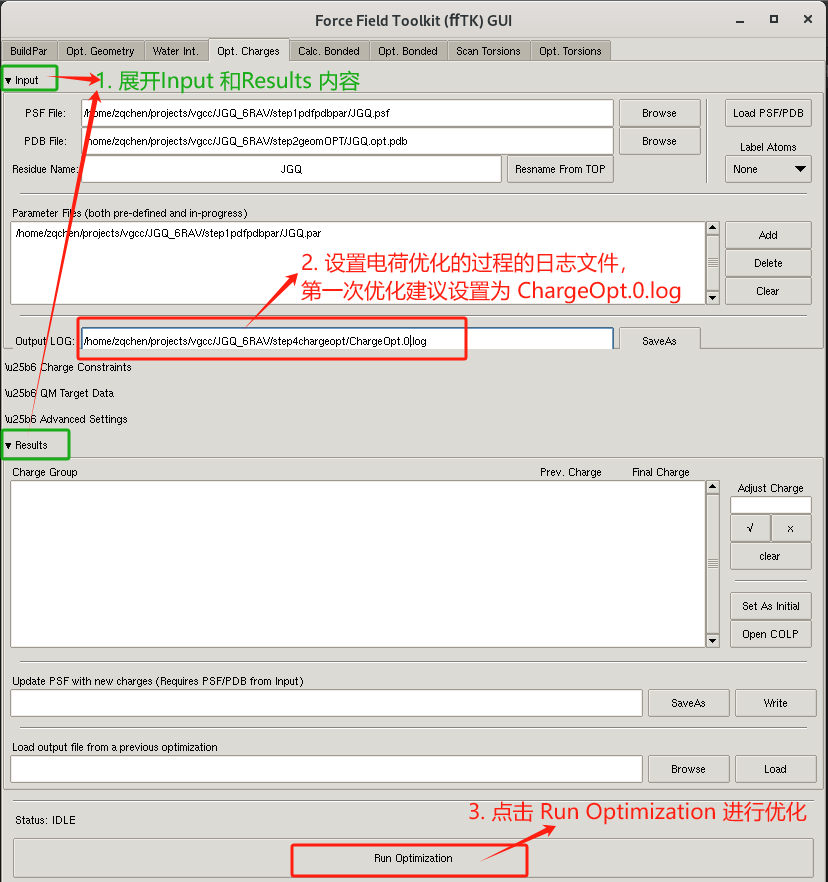
接着第一次对电荷进行优化
- 关闭charge constraints内容,关闭QM Target Data内容,打开Input和Results 内容。
- 设置电荷优化日志文件 ChargeOpt.0.log。
- 点击run optimization对电荷进行优化。
如下图所示:
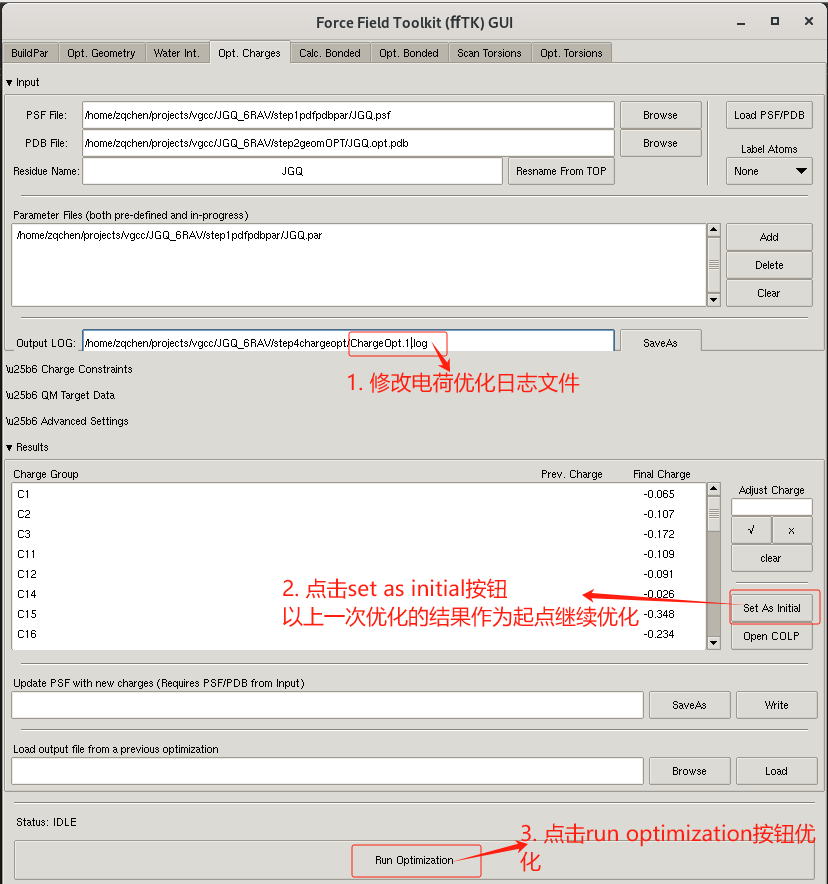
接着 迭代对电荷优化直到收敛
- 设置电荷优化日志文件 ChargeOpt.1.log。
- 点击set as initial 按钮,在上一次优化的基础上进行优化。
- 点击run optimization对电荷进行优化。
如下图所示:
- 设置电荷优化日志文件 ChargeOpt.2.log。
- 点击set as initial 按钮,在上一次优化的基础上进行优化。
- 点击run optimization对电荷进行优化。
重复迭代优化1-10次,直到收敛,电荷不再变化。
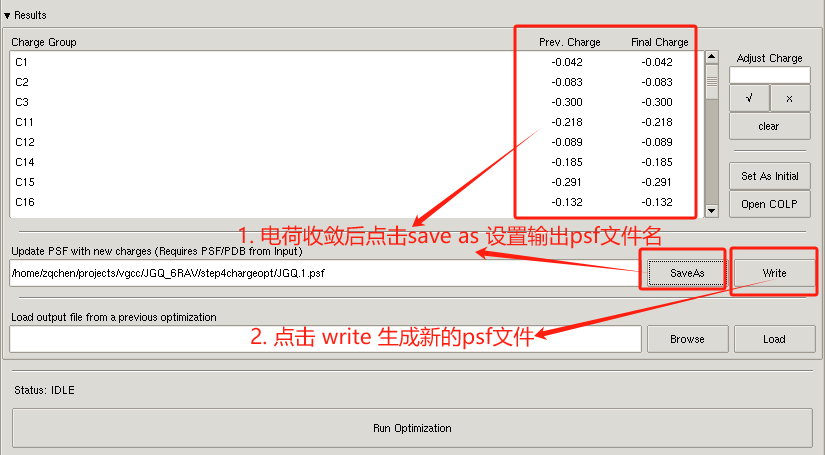
最后 保存结果到psf文件中
- 关闭其他内容,打开Results 内容。
- 点击save as 设置新的psf文件名 JGQ.1.psf 。
- 点击write按钮,生成JGQ.1.psf 文件。
如下图所示:
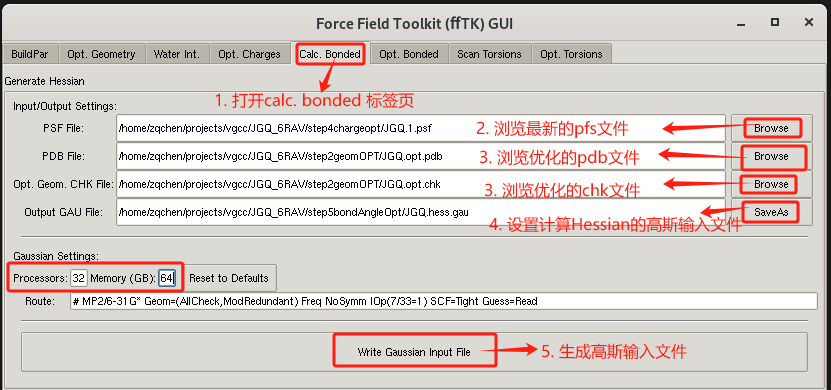
step3 键和角度优化¶
首先 准备计算Hessian矩阵的Gaussian输入文件
注解
计算配体和水分子相互作用能需要3个文件:1. 最新的psf文件;2. 结构优化后的pdb文件;3. 结构优化后的chk文件;
- 点击 Extensions-> Modeling -> Force Field toolkit 工具,打开fftk插件;选择Calc. Bonded标签页。
- 点击Browse 加载 psf 文件JGQ.1.psf。
- 点击Browse 加载结构优化后的 pdb 文件JGQ.opt.pdb。
- 点击Browse 加载结构优化后的 chk 文件JGQ.opt.chk。
- 点击save as 设置计算Hessian矩阵的输入文件名 JGQ.hess.gau。
- 设置Gaussian参数,并点击Write Gaussian Input Files生成Gaussian的输入文件。
如下图所示:
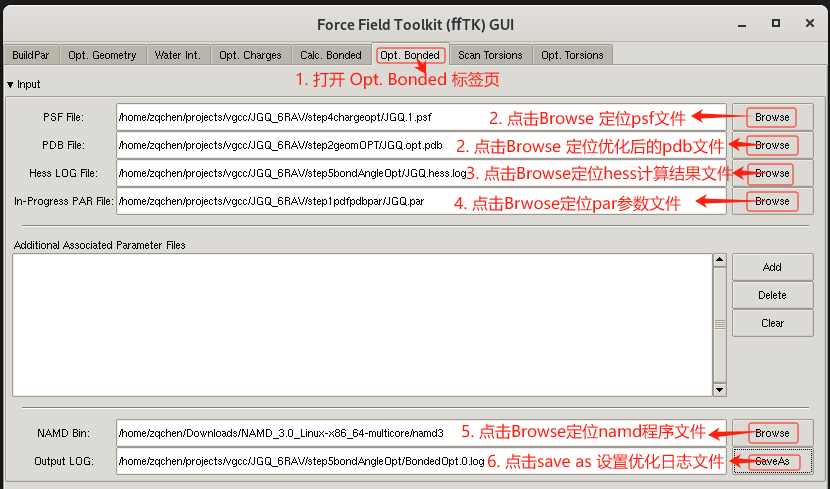
接着 执行量化计算
nohup g16 JGQ.hess.gau &
准备 Bond.opt 输入文件
- 点击 Extensions-> Modeling -> Force Field toolkit 工具,打开fftk插件;选择Opt. Bonded标签页,展开input内容。
- 点击Browse 加载 psf 文件JGQ.1.psf。
- 点击Browse 加载结构优化后的 pdb 文件JGQ.opt.pdb。
- 点击Browse 加载hess矩阵计算的结果文件JGQ.hess.log。
- 点击Browse 加载力场参数文件 JGQ.par。
- 点击Browse 配置namd程序的位置。
- 点击save as 设置计算Bond. opt的计算日志文件BondedOpt.0.log。
如下图所示:
注解
如果是用g16计算的JGQ.hess.log, 需要修改 JGQ.hess.log文件的内容。 删除 Redundant internal coordinates found in file. (old form). 这一行。 增加 Redundant internal coordinates taken from checkpoint file:
和JGQ.hess.chk这2句。
g09的hesslog文件格式如下:
Redundant internal coordinates taken from checkpoint file:
H49.hess.chk
Charge = 0 Multiplicity = 1
C,0,66.3033902412,94.7823683521,74.1186784493
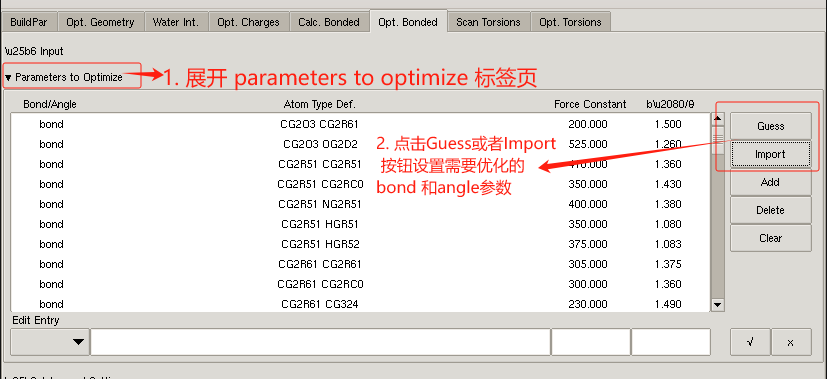
设置 需要优化的bond 和angle的参数
- 选择Opt. Bonded标签页,关闭input内容,打开Prameters to optimize内容进行优化键和键角参数。
- 设置键和键角的初始参数,有2种方法,方法1:通过Guess按钮进行设置;方法2:通过import按钮进行设置。
注解
Guess按钮是基于从par文件提取键和键角,并自动设置初始值。 Import按钮是从par文件中提取键和键角以及初始值。 可以2种方法都试试,哪种方法收敛小,用哪种方法。
如下图所示:
第一次 优化的bond 和angle的参数
- 选择Opt. Bonded标签页,打开Input内容和Result内容 。
- 设置优化日志文件BondedOpt.0.log 。
- 点击优化按钮Run Optimization 。
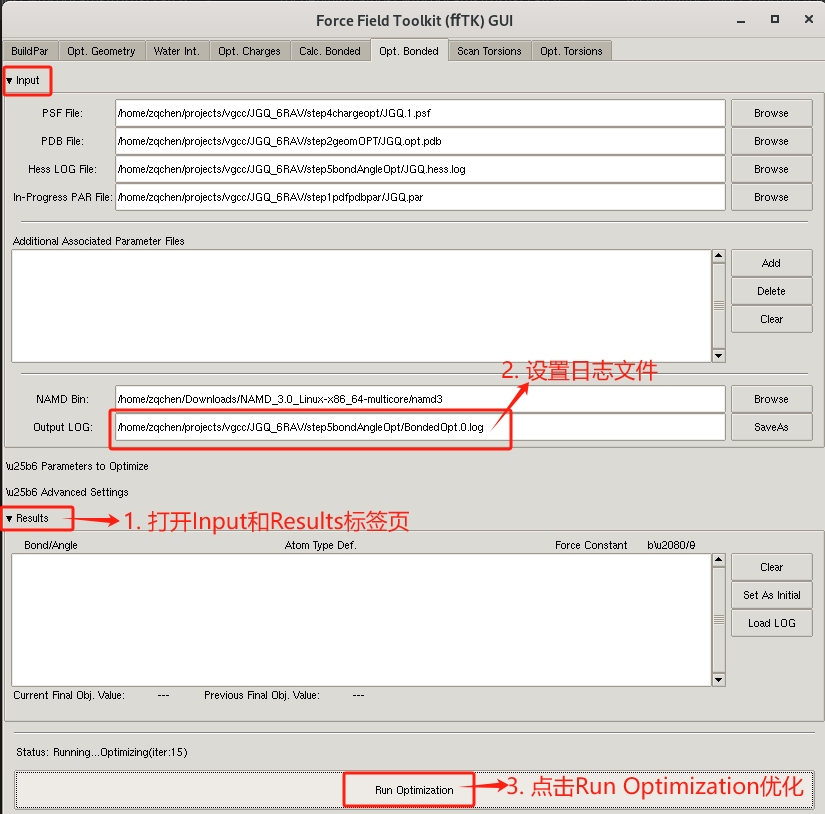
迭代优化
- 设置优化日志文件BondedOpt.n.log, n代表优化次数 。
- 点击set as initial按钮。
- 点击优化按钮Run Optimization 。
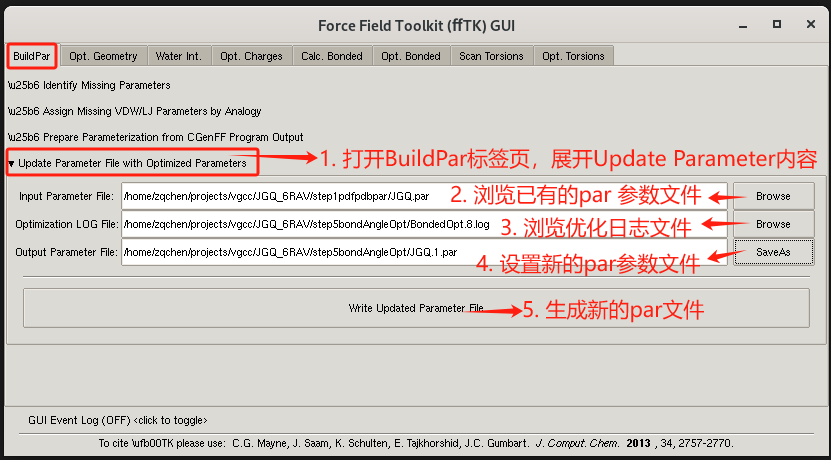
最后更新par参数
- 点击 Extensions-> Modeling -> Force Field toolkit 工具,打开fftk插件;选择BuildPar标签页,展开update parameter file with optimized parameters内容。
- 点击Browse 加载 par 文件JGQ.par 。
- 点击Browse 优化后日志文件BondedOpt.8.log。
- 点击save as 设置新的par文件 JGQ.1.par 。
- 点击Write Updated parameter file 更新优化后的参数到新的par文件 JGQ.1.par中。
如下图所示:
step4 二面角优化¶
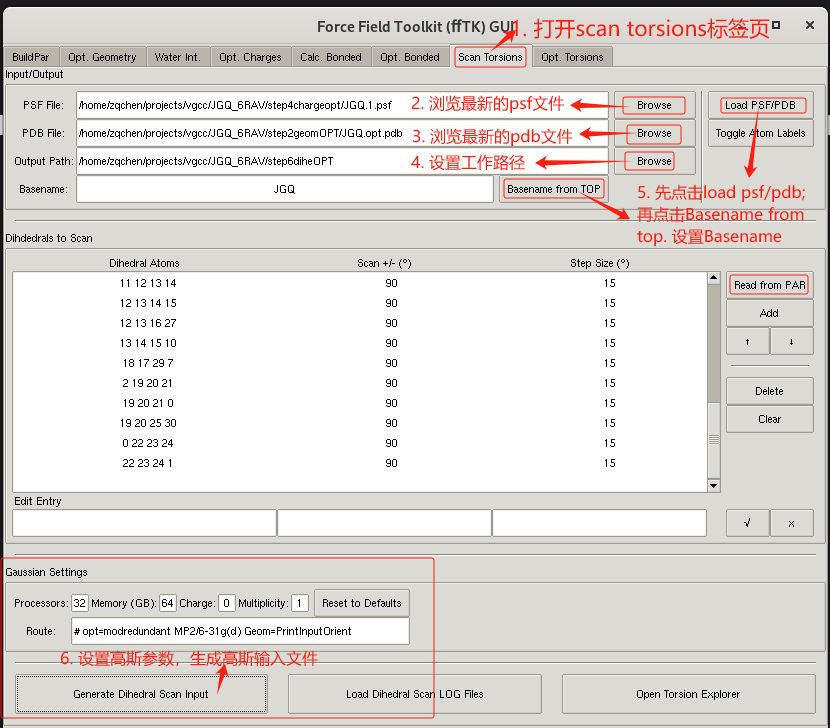
首先 准备二面角扫描的Gaussian输入文件
- 点击 Extensions-> Modeling -> Force Field toolkit 工具,打开fftk插件;选择 scan torsion 标签页。
- 点击Browse 加载最新的 psf 文件JGQ.1.psf。
- 点击Browse 加载结构优化后的 pdb 文件JGQ.opt.pdb。
- 点击Browse 设置二面角优化的工作输出路径。
- 点击load psf/pdb按钮,再点击Basename from top设置Basename。
- 点击Read from par设置需要优化的二面角。
- 设置Gaussian参数,并点击Generate Dihedral scan Input按钮生成Gaussian的输入文件。
如下图所示:
然后 进行量化计算
依次对所有文件进行逐一量化计算:
nohup g16 JGQ.scan*.gau &
计算结束后,检查log文件是否正常结束,对于不正常结束的log文件,分析原因,重新计算或者删除log文件。
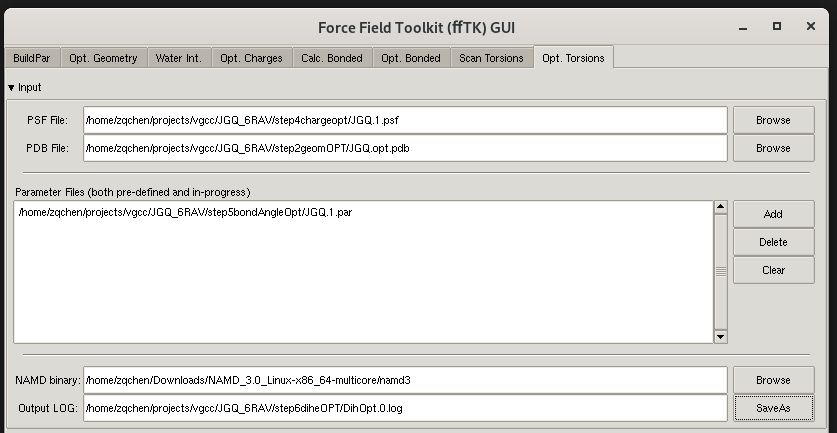
接着 优化二面角参数1–载入输入文件
- 点击 Extensions-> Modeling -> Force Field toolkit 工具,打开fftk插件;选择 Opt. torsion 标签页,展开input内容。
- 点击Browse 加载最新的 psf 文件JGQ.1.psf。
- 点击Browse 加载结构优化后的 pdb 文件JGQ.opt.pdb。
- 点击Add 加载优化后的参数文件JGQ.1.par。
- 点击Browse 设置NAMD程序的路径。
- 点击save as 设置二面角参数优化日志文件 DihOpt.0.log
如下图所示:
接着 优化二面角参数2–载入量化结果文件
- 关闭input内容,打开QM Target Data内容。
- 点击add 按钮 加载所有的量化结果log文件。
如下图所示:
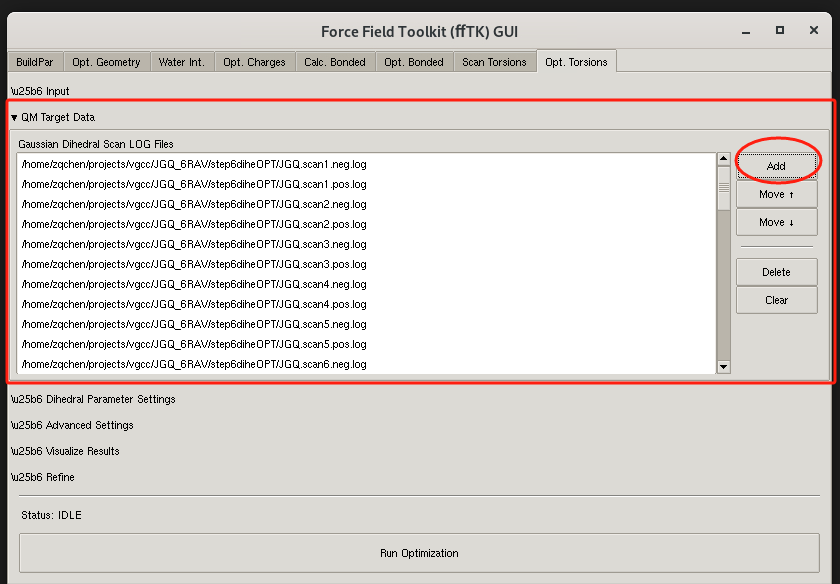
接着 优化二面角参数3–获取需要优化的二面角参数
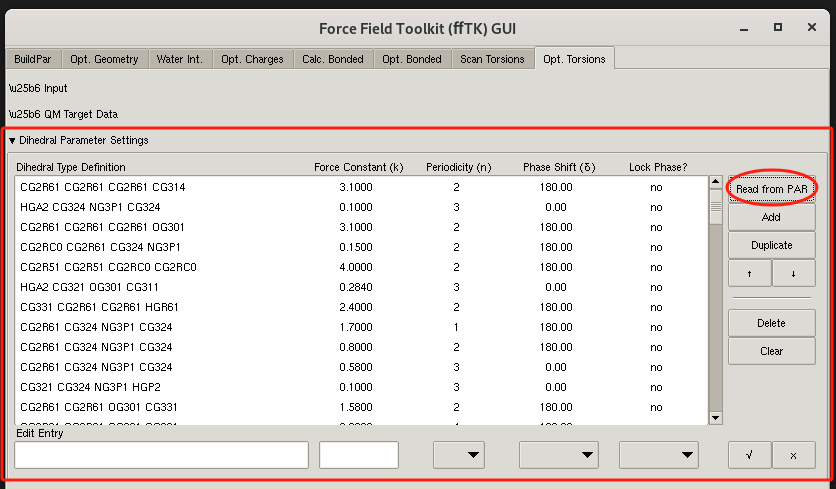
- 关闭input内容,关闭QM Target Data内容,打开Dihedral Parameter settings参数。
- 点击 Read From Par按钮加载优化后的参数文件JGQ.1.par,读取二面角参数。
如下图所示:
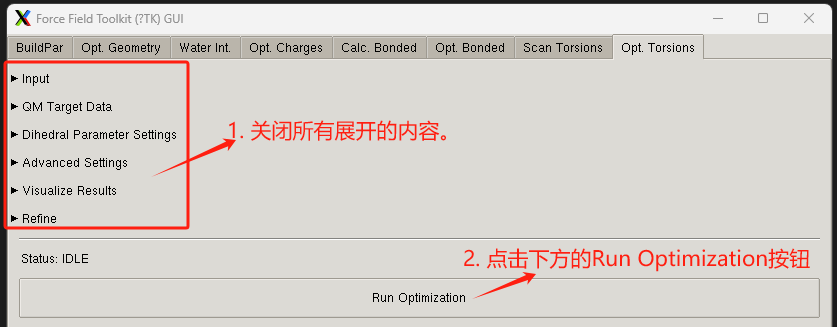
接着 优化二面角参数4–第一次优化二面角参数
- 关闭所有展开的内容。
- 点击下方的Run Optimization按钮,进行优化。
如下图所示:
注解
注意par文件中的参数格式,不能少0.00。 !atom types Kpsi psi0 ! CG2O3 OG2D2 OG2D2 CG2R61 96.0000 0.00 ! ! PROT 90.0->96.0 acetate, single impr (KK) WILDCARD
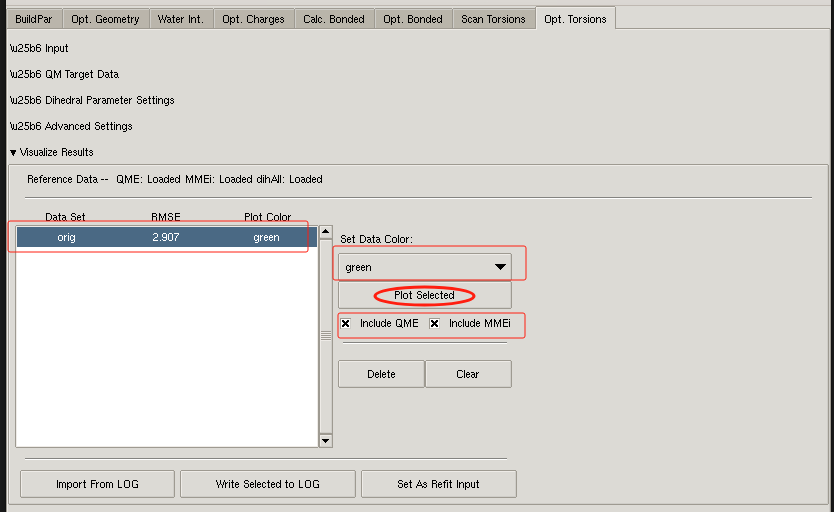
接着 优化二面角参数4–可视化QM和MM的结果
- 展开Visual Results内容;
- 选择orig条目;
- 设置MM结果的颜色;
- 确认勾选QME 和MMEi;
- 点击plot 可视化QM 和MM的结果。
如下图所示:
接着 优化二面角参数4–继续优化
- 展开Visual Results内容;
- 选择最新的优化结果;
- 点击set as initial input按钮。
如下图所示:
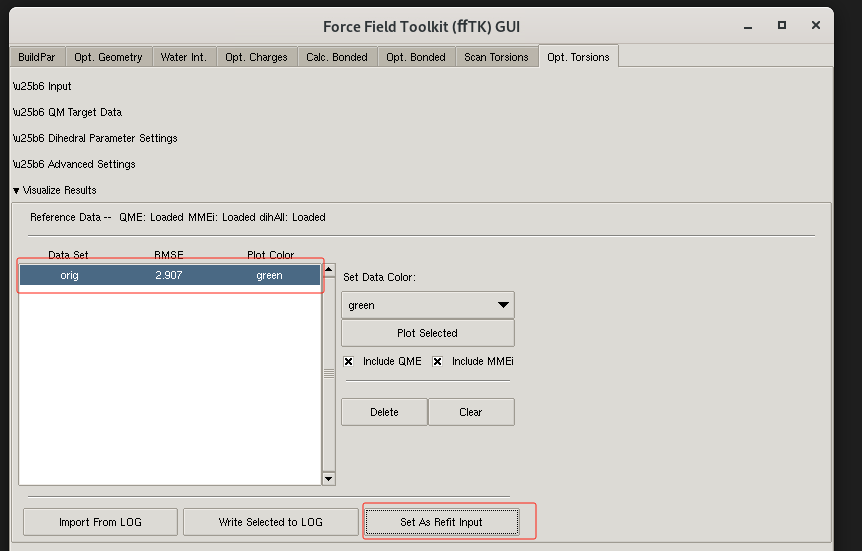
- 关闭Visual Results内容,展开refine内容,点击Run Refitting/Refinement 按钮。
如下图所示:
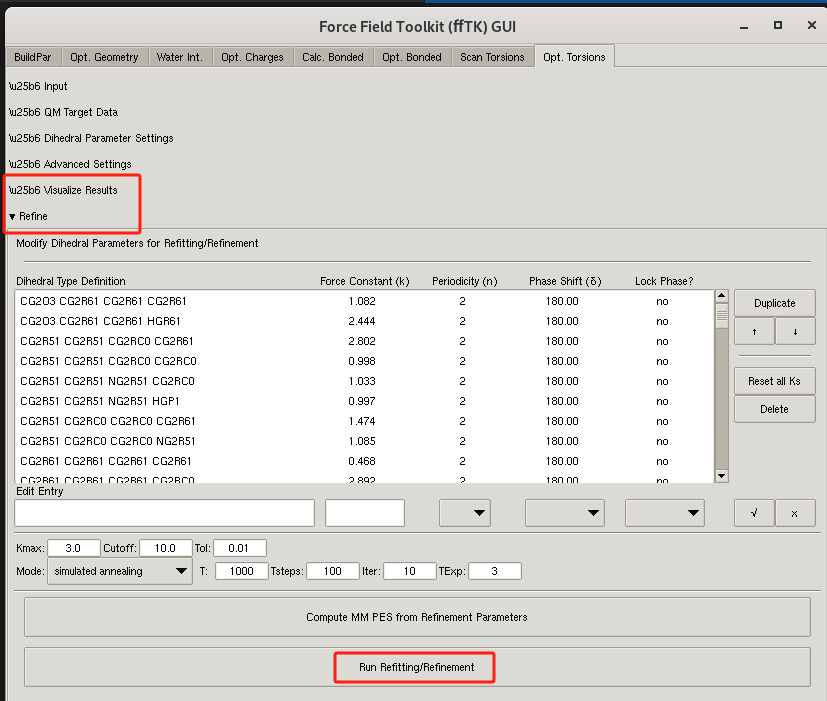
注解
重复优化,优化过程中可调节优化参数,比如优化方法和参数。
直到完成优化,QM和MM比较一致为止。 RMSE 越小越好。
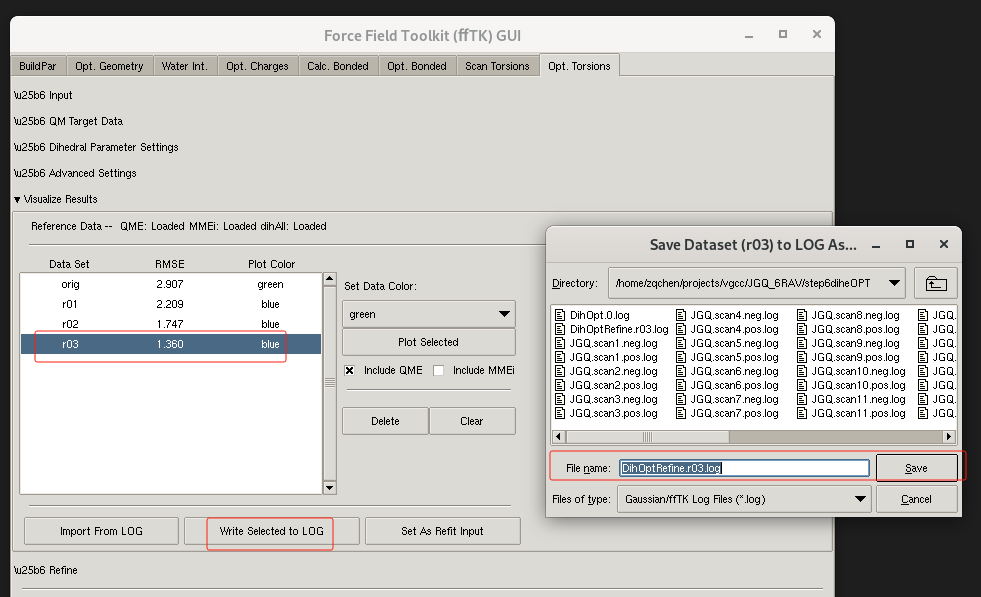
- 优化完成后,关闭refine,在visual results中选择最好的优化结果;
- 点击 write selected to log按钮写入log文件DihOptRefine.r3.log。
如下图所示:
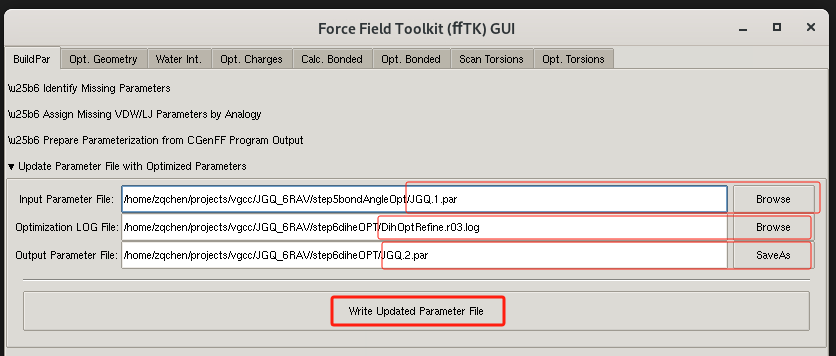
最后 更新par文件
- 点击 Extensions-> Modeling -> Force Field toolkit 工具,打开fftk插件;选择BuildPar标签页,展开update parameter file with optimized parameters内容。
- 点击Browse 加载 par 文件JGQ.1.par 。
- 点击Browse 优化后日志文件DihOptRefine.r03.log
- 点击save as 设置新的par文件 JGQ.2.par 。
- 点击Write Updated parameter file 更新优化后的参数到新的par文件 JGQ.1.par中。
如下图所示:
注解
基于psf 文件和par 文件更新str文件,就可以进行分子动力学模拟。
